You are here
Over klinische studies
Hier krijgt u begrijpbare en betrouwbare informatie over klinische studies. Klik op onderstaande vragen voor heldere antwoorden.
Waarom zijn klinische studies belangrijk?
Wie deelneemt aan een klinische studie, als patiënt of als gezonde vrijwilliger, helpt mee aan de vooruitgang van de geneeskunde en aan een betere toekomst voor patiënten.
Voor veel patiënten zijn bestaande geneesmiddelen niet geschikt of niet doeltreffend. Daarom voeren wereldwijd honderdduizenden wetenschappers onderzoek naar nieuwe geneesmiddelen en behandelingen. Deze kunnen echter niet op de markt gebracht worden zonder eerst klinische studies met gezonde vrijwilligers en patiënten uit te voeren. Deze klinische studies worden uitgevoerd om de veiligheid, de werkzaamheid en het effect op lange termijn te kennen. Daarnaast worden nieuwe geneesmiddelen of behandelingen ook vergeleken met bestaande.
Het is noodzakelijk dat voldoende personen deelnemen aan deze studies zodat wetenschappelijk onderbouwde conclusies kunnen genomen worden.
Wat is klinisch onderzoek?
Tijdens de ontwikkeling van een nieuw geneesmiddel moet men bewijzen dat dit nieuw geneesmiddel veilig is en dat het werkt alvorens men het op de markt kan brengen.
De ontwikkeling van geneesmiddelen wordt onderverdeeld in enerzijds pre-klinisch onderzoek met proefbuizen en dieren, en anderzijds in klinisch onderzoek met mensen. Pas als aangetoond is dat het nieuw product veilig en werkzaam is bij proefdieren zal het onderzocht worden bij mensen.
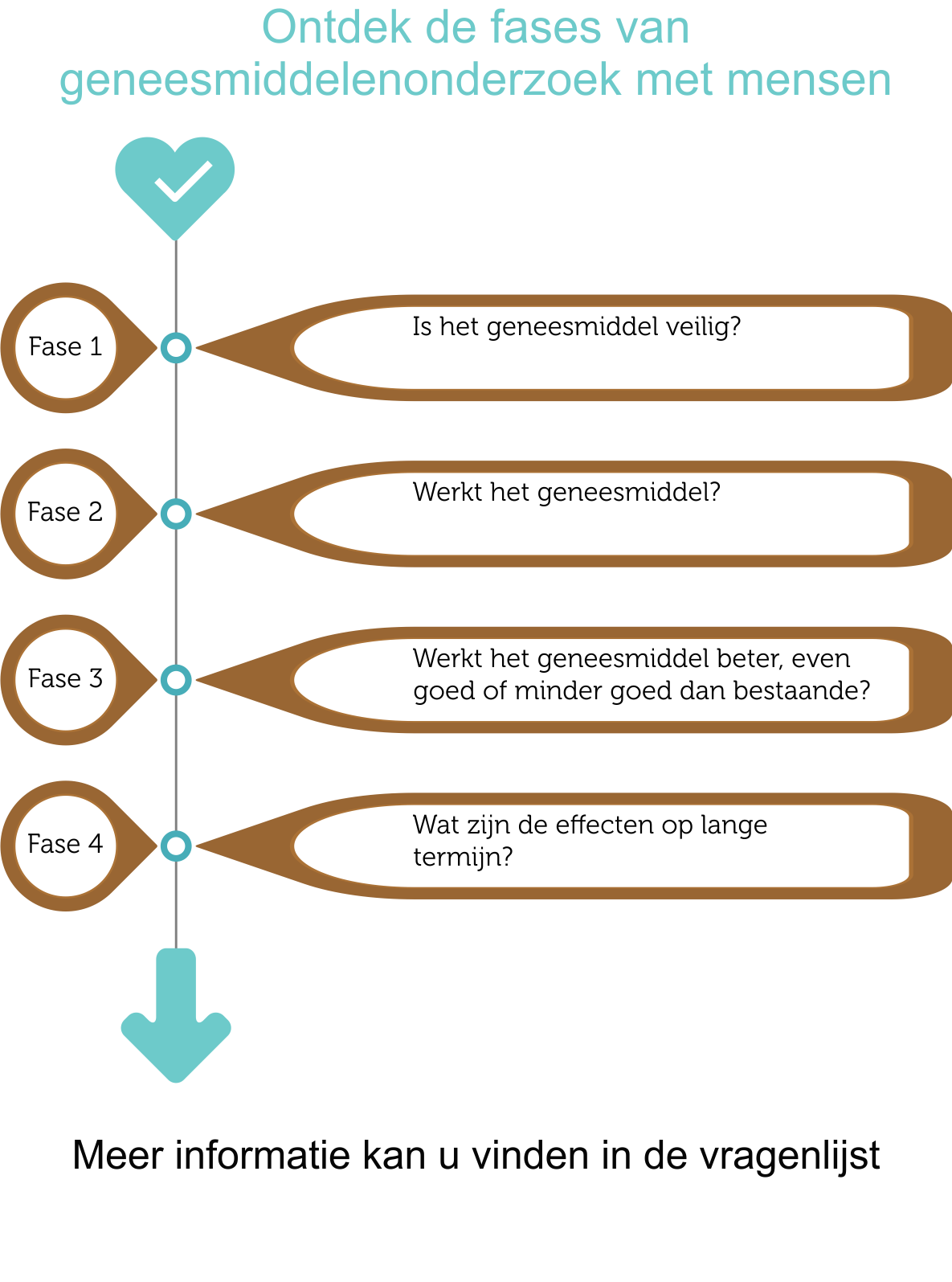
De testen die worden uitgevoerd bij mensen om na te gaan of het nieuw product veilig en werkzaam is, worden klinische studies genoemd. Deze studies worden uitgevoerd in een set van 4 opeenvolgende fases (I, II, III en IV). De doelstelling van een klinische studie is afhankelijk van het nieuw product dat wordt onderzocht.
Wat zijn de fases van klinisch onderzoek bij mensen?
- Pre-klinisch onderzoek met proefbuizen en proefdieren
- Klinisch onderzoek met mensen
Klinisch onderzoek bestaat uit streng gecontroleerde klinische studies in 4 opeenvolgende fasen.
- Fase 1 studies – Is het geneesmiddel veilig?
Het potentieel nieuw geneesmiddel wordt meestal toegediend aan een kleine groep (20 tot 100) gezonde vrijwilligers om de algemene effecten (verdeling, metabolisme en uitscheiding, alsook eventuele ongewenste bijwerkingen) van het nieuw geneesmiddel te bestuderen. - Fase 2 studies – Werkt het geneesmiddel?
Wanneer uit de resultaten van de fase I studie kan worden vastgesteld dat het potentieel nieuwe geneesmiddel veilig is, wordt een fase II studie opgestart bij een kleine groep geselecteerde patiënten (100 tot 500) die op vrijwillige basis deelnemen. Tijdens deze fase gaat men na of het potentieel nieuw geneesmiddel werkt voor de ziekte waarvoor het ontwikkeld werd. De patiënten die kunnen deelnemen worden geselecteerd op basis van strikte in- en exclusie criteria. Dit doet men om de variatie binnen de groep te minimaliseren zodat wetenschappelijk onderbouwde conclusies kunnen genomen worden. - Fase 3 studies - Werkt het geneesmiddel beter, even goed of minder goed dan bestaande geneesmiddelen of behandelingen?
Het potentieel nieuw geneesmiddel wordt onderzocht in een grotere groep patiënten (1000 tot 5000) om de doeltreffendheid en de veiligheid op langere termijn te bestuderen. In deze fase wordt het geneesmiddel vergeleken met bestaande geneesmiddelen of behandelingen. Als kan aangetoond worden dat het geneesmiddel voldoende of een beter resultaat heeft en veilig is, wordt het officieel geregistreerd. Vanaf dat moment is het nieuw geneesmiddel op de markt verkrijgbaar en kan het voorgeschreven worden door artsen. - Fase 4 studies – Wat zijn de effecten op lange termijn?
Ook na de introductie van het nieuw geneesmiddel op de markt, wordt de veiligheid verder opgevolgd. Tijdens fase IV studies wordt vooral gekeken naar mogelijke bijwerkingen die zich sporadisch voordoen en naar de complicaties op lange termijn. Daarnaast wordt soms ook onderzocht of het geneesmiddel werkzaam is voor andere ziektes of aandoeningen.
Wie voert klinische studies uit?
Er is een onderscheid tussen (i) commerciële studies en (ii) niet-commerciële of academische studies. De initiatiefnemer van een commerciële studie is een farmaceutisch bedrijf of een bedrijf dat medische hulpmiddelen ontwikkelt. Bij een academische studie is de initiatiefnemer een onderzoeksarts uit een ziekenhuis.
Beide soorten studies moeten aan dezelfde strenge regels voldoen. In beide gevallen is een proefpersoon dus even goed beschermd en heeft de proefpersoon dezelfde rechten en plichten.
Bij commerciële studies is er geen rechtstreeks contact tussen de patiënten en de initiatiefnemer. Alle contacten verlopen via een deelnemend onderzoekscentrum en een onderzoeksarts verbonden aan de studie. De onderzoeksarts zorgt voor begeleiding, opvolging en onderzoek van de proefpersonen.
Wie kan deelnemen aan een klinische studie?
Zowel gezonde vrijwilligers als patiënten kunnen bij een studie betrokken zijn.
Elke studie heeft specifieke voorwaarden en deze worden steeds beschreven in het studieprotocol. Een protocol van een klinische studie omvat alle regels en procedures van een studie. Het is als het ware het referentieboek/draaiboek voor het correct uitvoeren van een studie. Het protocol moet steeds goedkeuring krijgen van een Ethisch Comité en van de overheid alvorens de studie kan starten.
De voorwaarden tot deelname worden ook wel de inclusie en exclusie criteria genoemd.
Mogelijke deelnemers:
- moeten voldoen aan de studiespecifieke voorwaarden;
- zullen en moeten uitgebreid geïnformeerd zijn aan de hand van het informatieformulier;
- moeten begrijpen wat deelname betekent, inclusief wat de mogelijke risico’s en voordelen zijn;
- moeten enkel op vrijwillige basis kunnen deelnemen zoals uitgelegd in het informatieformulier;
- moeten verzekerd zijn tegen mogelijke schade;
- moeten op elk moment kunnen stoppen;
- moeten instemmen en een document, het zogenaamde informatie- en toestemmingsformulier, ondertekenen voor aanvang van de onderzoeksprocedures.
Is deelnemen aan een klinische studie veilig en wie garandeert deze veiligheid?
Klinische studies zijn zeer streng gereglementeerd om het risico voor de deelnemende proefpersonen te beperken tot een minimum. Om de rechten, de integriteit, de confidentialiteit van de proefpersonen, en de geloofwaardigheid van de studiegegevens te beschermen, werden de internationale ethische en wetenschappelijke voorwaarden gedefinieerd in de Declaratie van Helsinki en de Guideline of Good Clinical Practice (GCP).
Voor de start van een klinische studie, moet eerst een goedkeuring verkregen worden van een erkend Ethisch Comité en de toelating van de overheid (in België is dit het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten, kortweg FAGG). Beide voorvermelde instanties spreken een advies uit en beoordelen of het nieuwe geneesmiddel veilig is en of de voordelen van het nieuwe geneesmiddel opwegen tegen de eventuele risico’s verbonden aan de studie. Pas na het verkrijgen van een gunstig advies (= goedkeuring) kan de studie van start gaan.
Het bevoegde Ethisch Comité blijft gedurende de volledige duur van de studie waken over de belangen van de deelnemers. Op elk moment kan beslist worden om de studie stop te zetten, bijvoorbeeld omwille van onaanvaardbare bijwerkingen.
Wat zijn de mogelijke risico’s van een deelname aan een klinische studie?
Eerst wordt een studieprotocol opgesteld voor het uitvoeren van een klinische studie. Een protocol van een klinische studie omvat alle regels van een studie. Het is als het ware het referentieboek/draaiboek voor het uitvoeren van een studie. Het protocol moet steeds goedkeuring krijgen van een Ethisch Comité en de overheid alvorens de studie kan starten en ook tijdens de studie blijft het Ethisch Comité waken over de belangen van de deelnemers.
Desondanks kunnen er risico’s verbonden zijn aan de deelname van een klinische studie of kan het enkele nadelen met zich meebrengen. Het risicoprofiel van een studie wordt vooraf geanalyseerd door verschillende partijen (opdrachtgevende firma, artsen, Ethische Comités en de overheid) en enkel indien de voordelen opwegen tegenover de mogelijke risico’s kan een studie van start gaan.
Indien men spreekt over risico’s/nadelen, wordt gedacht aan:
- Het geneesmiddel of de behandelingstherapie is meestal nieuw en wordt nog onderzocht. Dit betekent vaak dat nog niet alle bijwerkingen of nevenwerkingen gekend zijn. Het risico op ongekende bijwerkingen of nevenwerkingen is verschillend per studie en is niet altijd volledig te voorspellen. Daarom zal de onderzoeksarts de proefpersoon hierover vooraf meer informatie geven. Uiteraard worden de bijwerkingen en nevenwerking van dichtbij opgevolgd door zowel de onderzoeksarts als door de ethische comités die waken over de veiligheid van alle deelnemers. Daarenboven kan een deelnemer ook op ieder ogenblik beslissen om te stoppen met een studie.
- Het is mogelijk dat het nieuwe geneesmiddel geen toegevoegde waarde heeft.
- Meestal weet de deelnemer bij fase II en fase III klinische studies niet of hij/zij het nieuwe geneesmiddel wel of niet krijgt. Indien het ethisch verantwoord is, kan het studieprotocol voorzien dat er een controlegroep is die geen actief product krijgt, maar een placebo.
- Deelname kan een extra inspanning vergen van de proefpersoon, zoals extra ziekenhuisbezoeken, extra onderzoeken of procedures zoals bijvoorbeeld extra bloedafnames,...
Vragen over uw mogelijke deelname
Overweegt u om zich aan te melden voor een klinische studie? Klik op onderstaande vragen met betrekking tot uw mogelijke deelname.
Waarom zou u deelnemen en wat zijn de mogelijke voordelen van uw deelname aan een klinische studie?
- Door deel te nemen aan een klinische studie ondersteunt u het medisch onderzoek. U helpt hierdoor mee aan de vooruitgang van de geneeskunde.
- Door als patiënt deel te nemen kan u genieten van de meest recente gezondheidszorg en van een mogelijk nieuwe behandeling voor uw ziekte waarvan de doeltreffendheid en veiligheid onderzocht worden.
- De resultaten van de studies kunnen ook toekomstige patiënten helpen door de ontwikkeling van een potentieël nieuw geneesmiddel, een nieuw vaccin, een betere diagnostische test, ...
- Het is mogelijk dat de onderzoeksarts meer testen doet dan wat hij/zij normaal zou doen en dit om de effecten van het toegediende geneesmiddel zo goed mogelijk op te volgen. Deze bijkomende behandelingen en/of testen tijdens de studie worden u niet aangerekend.
- De studies zijn strikt vertrouwelijk en uw privacy komt op geen enkel moment in het gedrang.
Er is evenwel geen enkele garantie dat uw deelname u een persoonlijk voordeel zal opleveren. U dient ook rekening te houden met de extra inspanningen en risico’s die een studie mogelijk met zich meebrengt. De mogelijke voordelen, maar ook de nadelen en de mogelijke risico’s van een specifieke studie zullen steeds vooraf uitvoerig worden uitgelegd en besproken door uw onderzoeksarts.
Wie beslist of u kan deelnemen aan een klinische studie?
De onderzoeksarts oordeelt of u kan deelnemen aan een klinische studie.
Deelnemen aan een klinische studie is steeds geheel vrijwillig. Als u beslist om niet deel te nemen aan de studie, zal dit in geen enkel geval invloed hebben op uw verdere behandeling en zal dit ook uw relatie met uw behandelende arts niet beïnvloeden. U kan ook beslissen om op eender welk ogenblik te stoppen met de studie.
Meer details over onze rol vindt u in ons doorverwijsbeleid.
Wat gebeurt er met uw gegevens tijdens een klinische studie?
Als u voldoet aan de selectieciteria en besluit om deel te nemen aan een klinische studie, worden uw deelname en al uw persoonlijke en medische gegevens vertrouwelijk behandeld in overeenstemming met de lokale wetten. Tijdens een klinische studie worden uw gegevens op een gecodeerde wijze verzameld en behandeld. Deze gegevens worden uitsluitend verwerkt en geanalyseerd in een wetenschappelijke context die samenhangt met de studie. In latere wetenschappelijke publicaties van de studieresultaten wordt geen informatie meegedeeld waaruit de identiteit van de deelnemers kan worden afgeleid.
Uw onderzoeksarts zal op geen enkel moment medische gegevens die worden verzameld tijdens een klinische studie delen met C-Lys. C-Lys is immers enkel betrokken in de voorselectie van kandidaat proefpersonen.
Wat gebeurt er indien u schade zou oplopen tijdens een studie?
Iedere klinische studie is zorgvuldig opgesteld om het risico tot een minimum te beperken. Onvoorziene problemen zijn echter niet uit te sluiten. Daarom stelt de wetgeving dat de initiatiefnemer van de studie verplicht is om voor elke klinische studie een verzekering af te sluiten ter bescherming van de deelnemers. C-Lys is enkel betrokken in de voorselectie van kandidaat proefpersonen en is niet verantwoordelijk voor uw deelname aan de studie. Voor meer informatie over de verzekering kan u bij uw onderzoeksarts van de studie terecht.
Als u geïnteresseerd bent om deel te nemen aan een klinische studie, kan een onderzoeksarts u het doel van de studie en mogelijke voordelen uitleggen. Uw arts zal u er ook op wijzen dat deze niet gegarandeerd kunnen worden. Er kunnen altijd ongewenste bijwerkingen optreden omwille van het onderzoeksstadium waarin het nieuwe geneesmiddel of de behandeling zich nog bevindt.
Krijgt u een financiële vergoeding voor uw deelname aan een klinische studie?
Voor een deelname aan fase I studies als gezonde vrijwilliger wordt meestal een financiële vergoeding voorzien die berekend wordt op basis van de belasting van de onderzoeken (aantal bloedafnames, aantal overnachtingen...) die u ondergaat voor de studie. Voor een deelname aan fase II, III en IV studies als patiënt is het wettelijk meestal niet toegestaan om een financiële vergoeding te ontvangen. U kan in sommige gevallen wel recht hebben op een onkostenvergoeding voor uw parking-en verplaatsingskosten. In beide gevallen zal de onderzoeksarts van de studie u hierover meer informatie kunnen geven.
Een financiële vergoeding of onkostenvergoeding gaat steeds uit van de initiatiefnemer van de studie. Aangezien C-Lys enkel betrokken is bij de voorselectie van kandidaat proefpersonen is C-Lys in geen geval verantwoordelijk voor het betalen van een vergoeding aan proefpersonen.
Waar moet u eventuele bijwerkingen melden?
Wanneer men deelneemt aan een klinische studie, is het uiterst belangrijk om eventuele bijwerkingen of neveneffecten te melden aan de behandelende arts. Dit helpt om de veiligheid van het geneesmiddel of de behandeling te controleren.
Deze C-Lys website is niet bedoeld voor het melden van mogelijke bijwerkingen. Voor klinische studies is een strenge regelgeving van toepassing ter bescherming van de deelnemers. Door deze unieke juridische situatie kunnen en mogen C-Lys medewerkers niet antwoorden op vragen over medische producten, medische behandelingen, bijwerkingen, of specifieke klinische studies. We beschikken enkel over de informatie die u kan terugvinden op deze website. De informatie over specifieke studies werd opgesteld in overleg met de onderzoekers en werd goedgekeurd door een bevoegde ethische commissie.
Deze C-Lys website is ook niet bedoeld om uw ervaring binnen een klinische studie te delen. Het kan verleidelijk zijn om te praten met anderen die net zoals u deelnemen aan een bepaalde klinische studie. Zo zou u over mogelijke bijwerkingen of uw ervaring kunnen vertellen, maar het is belangrijk om dit niet te doen. Het zou immers de uitkomst van het onderzoek kunnen beïnvloeden. Er bestaat namelijk zoiets als de kracht van de suggestie; horen over een neveneffect kan genoeg zijn om het ook echt op gang te brengen.
Klinisch onderzoek: van fase 1 tot fase 4.