You are here
Sobre estudios clínicos
Aquí encontrará información comprensible y fiable sobre estudios clínicos. Haga clic en las preguntas siguientes para obtener respuestas claras.
¿Por qué son importantes los estudios clínicos?
Las personas que participan en un estudio clínico, como pacientes o como voluntarios sanos, contribuyen al progreso de la medicina y a un futuro mejor para los pacientes.
Los medicamentos existentes no son aptos o son insuficientes para muchos pacientes. Por esta razón, miles de científicos por todo el mundo llevan a cabo investigaciones para desarrollar medicamentos y tratamientos nuevos. Sin embargo, éstos no se pueden comercializar sin antes haber efectuado estudios clínicos con pacientes y con voluntarios sanos. Estos estudios clínicos se realizan para conocer la seguridad, la eficacia y los efectos a largo plazo. Además, se comparan medicamentos o tratamientos nuevos con los ya existentes.
Es necesario un número suficiente de participantes en estos estudios para que se puedan presentar conclusiones con base científica.
¿Qué es la investigación clínica?
Durante la fase de desarrollo de un nuevo medicamento hace falta probar que el nuevo medicamento es seguro y que funciona, antes de su comercialización.
El desarrollo de medicamentos nuevos se divide en la investigación preclínica con tubos de ensayo y animales, por una parte, y la investigación clínica con sujetos humanos, por otra. Solo cuando se haya demostrado que el nuevo producto es seguro y eficaz en animales de laboratorio, empezarán los experimentos con seres humanos.
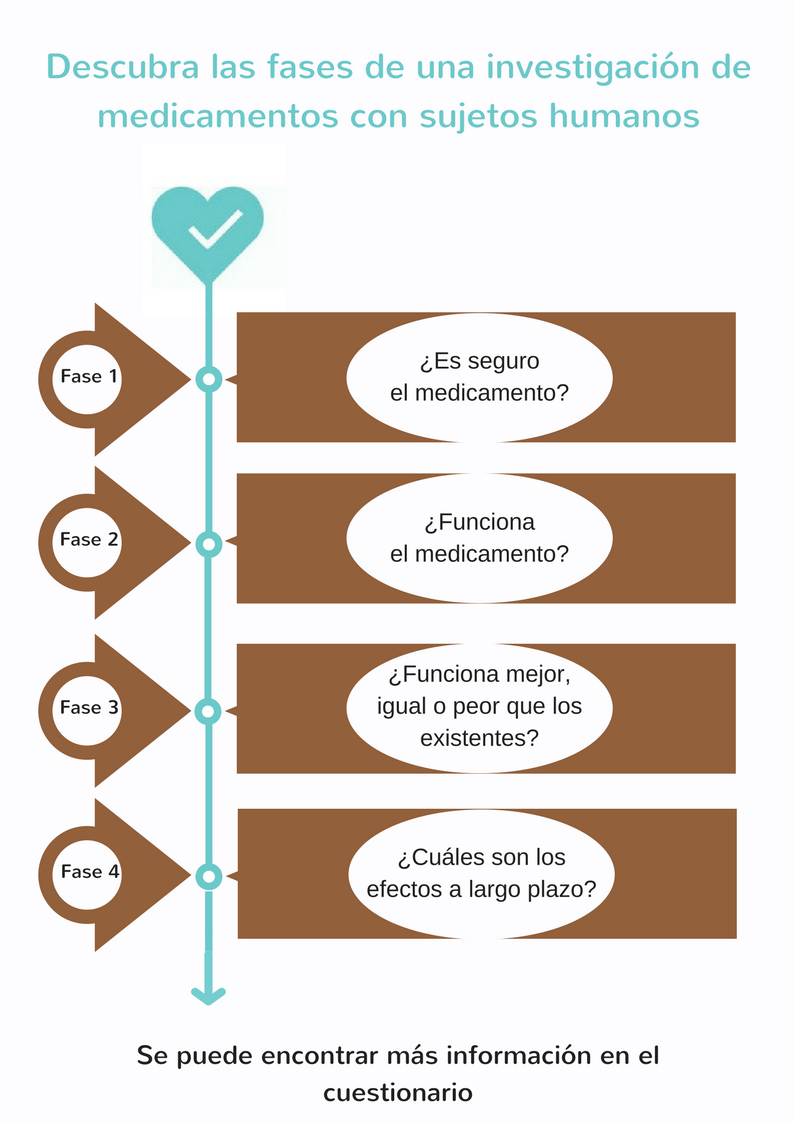
Los ensayos que se realizan con sujetos humanos para probar si un nuevo producto es seguro y eficaz se llaman estudios clínicos. Los estudios se realizan en una serie de 4 fases consecutivas (I, II, III y IV). El objetivo de un estudio clínico depende del nuevo producto que se investiga.
¿Cuáles son las fases de un estudio clínico con sujetos humanos?
- Investigación preclínica con tubos de ensayo y animales de laboratorio
- Investigación clínica con sujetos humanos
La investigación clínica consiste en estudios clínicos estrictamente supervisados en 4 fases consecutivas.
- Estudios de fase 1 – ¿Es seguro el medicamento?
Generalmente, se administra el nuevo medicamento potencial a un grupo reducido de voluntarios sanos (de 20 a 100) para investigar los efectos generales (repartición, metabolismo y excreción, así como los posibles efectos secundarios no deseados) del nuevo medicamento. - Estudios de fase 2 – ¿Funciona el medicamento?
Cuando los resultados del estudio de fase I demuestran que el nuevo medicamento potencial es seguro, se comienza un estudio de fase II con un grupo reducido de pacientes seleccionados (de 100 a 500) que participan voluntariamente. En esta fase, se investiga si el nuevo medicamento potencial es eficaz contra la enfermedad para la que ha sido desarrollado. La selección de los pacientes que participan se realiza a base de criterios estrictos de inclusión y exclusión. Así, se minimiza la variación dentro del grupo para poder llegar a conclusiones con base científica. - Estudios de fase 3 – ¿Funciona mejor, igual o peor que los medicamentos o los tratamientos existentes?
Se investiga el nuevo medicamento potencial en un grupo de pacientes mayor (de 1000 a 5000 participantes) para estudiar su eficacia y seguridad a largo plazo. En esta fase se compara el medicamento con medicamentos o tratamientos existentes. El nuevo medicamento se registrará oficialmente cuando se demuestre que da un resultado suficiente o mejor y que es seguro. A partir de entonces, el nuevo medicamento estará disponible en el mercado y los médicos pueden prescribirlo. - Estudios de fase 4 - ¿Cuáles son los efectos a largo plazo?
Se sigue controlando la seguridad del nuevo medicamento, incluso después de su introducción en el mercado. En los estudios de la fase IV se observan especialmente los efectos secundarios poco frecuentes y las complicaciones a largo plazo. A veces, también se investiga si el medicamento es eficaz para otras enfermedades o afecciones.
¿Quién realiza estudios clínicos?
Hay una distinción entre (i) estudios comerciales y (ii) estudios no comerciales o académicos. El promotor de un estudio comercial es una empresa farmacéutica o una empresa que desarrolla productos sanitarios. El promotor de un estudio académico es un médico de investigación de un hospital.
Ambos tipos de estudio deben cumplir las mismas reglas estrictas. En ambos casos la protección del sujeto de ensayo es del mismo nivel y el sujeto de ensayo tiene los mismos derechos y obligaciones.
En estudios comerciales no hay contacto directo entre los pacientes y el promotor. Cualquier contacto se establece a través del centro de investigación participante y del médico de investigación relacionado con el estudio. El médico de investigación se encarga del acompañamiento, el seguimiento y la investigación de los sujetos de ensayo.
¿Quién puede participar en un estudio clínico?
Tanto voluntarios sanos como pacientes pueden formar parte de un estudio.
Cada estudio tiene condiciones específicas que están definidas en el protocolo del estudio. El protocolo de un estudio clínico incluye todas las normas y los procedimientos de un estudio. Funciona como una guía de referencia/un plan de trabajo para llevar a cabo el estudio correctamente. El protocolo siempre debe ser aprobado por el comité de ética y el gobierno antes de que se pueda empezar con el estudio.
Las condiciones de participación también se suelen denominar los criterios de inclusión y exclusión.
Posibles participantes:
- deben cumplir las condiciones específicas del estudio;
- serán y deben ser informados exhaustivamente mediante el formulario de información;
- tienen que entender qué significa la participación, incluyendo los posibles riesgos y beneficios;
- solo pueden participar voluntariamente, tal y como se describe en el formulario de información;
- tienen que estar asegurados frente a posibles daños;
- deben poder dejar de participar en cualquier momento;
- tienen que dar su consentimiento y firmar un documento, el llamado formulario de información y consentimiento, antes de que se empiece con los procedimientos de investigación.
¿Es segura la participación en un estudio clínico y quién garantiza esta seguridad?
Los estudios clínicos están sujetos a una regulación muy estricta para minimizar el riesgo de los sujetos de ensayo participantes. Con el fin de proteger los derechos, la integridad y la confidencialidad de los sujetos de ensayo, y la credibilidad de los datos de estudio se han definido las condiciones éticas y científicas internacionales en la Declaración de Helsinki y en el Guideline of Good Clinical Practice (GCP).
Antes de que se pueda empezar un estudio clínico, se necesita la aprobación de un comité de ética autorizado y la autorización del gobierno (en Bélgica la Agencia Federal de Medicamentos y Productos Sanitarios – Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten, FAGG). Ambos órganos pronuncian sus consideraciones y evalúan si el nuevo medicamento es seguro y si los beneficios del nuevo medicamento compensan los posibles riesgos relacionados con el estudio. Solo se puede empezar con el estudio después de la obtención de un dictamen favorable (= aprobación).
El comité de ética autorizado sigue velando por los intereses de los participantes durante todo el estudio. Se puede decidir, en cualquier momento, que se suspende el estudio, por ejemplo, debido a efectos secundarios inaceptables.
¿Cuáles son los posibles riesgos relacionados con la participación en un estudio clínico?
Primero se define un protocolo según el cual se realizará el estudio clínico. Un protocolo de un estudio clínico incluye todas las normas del estudio. Funciona como una guía de referencia/un plan de trabajo para llevar a cabo el estudio correctamente. El protocolo siempre debe ser aprobado por el comité de ética y el gobierno antes de que se pueda empezar con el estudio. También durante el estudio el comité de ética sigue velando por los intereses de los participantes.
Sin embargo, siempre puede haber riesgos asociados a la participación en un estudio clínico o posibles desventajas. El perfil de riesgo de un estudio es analizado previamente por varias partes (la empresa ordenante, médicos, los comités de ética y el gobierno) y, únicamente, cuando los beneficios compensan los posibles riesgos se puede empezar con el estudio.
Los posibles riesgos/desventajas incluyen:
- El medicamento o el tratamiento suele ser nuevo y está aún siendo examinado. Esto significa, generalmente, que todavía no se conocen todos los efectos no deseados o efectos secundarios. El riesgo de efectos no deseados o secundarios es diferente para cada estudio y no siempre es completamente previsible. Por lo tanto, el médico del estudio informará bien al sujeto de ensayo sobre este asunto antes de empezar. Por supuesto, tanto el médico como los comités de ética, que velan por la seguridad de los participantes, seguirán muy de cerca los efectos no deseados y los efectos secundarios. Asimismo, el participante tiene la libertad de abandonar el estudio en cualquier momento.
- Es posible que el nuevo medicamento no tenga un valor añadido.
- Generalmente, el participante de un estudio clínico de fase II o III no sabe si recibe el nuevo medicamento o no. Si está justificado desde el punto de vista ético, el protocolo de estudio puede prever un grupo de control que no recibe un producto activo, sino un placebo.
- La participación puede exigir un esfuerzo extra del sujeto de ensayo, tales como consultas adicionales en los hospitales, revisiones médicas o procedimientos adicionales, por ejemplo, análisis de sangre, etc.
Preguntas sobre su posible paricipación
¿Usted considera registrarse para un estudio clínico? Haga clic en las preguntas siguientes asociadas con su posible participación.
¿Por qué participaría usted y cuáles son los posibles beneficios de su participación en un estudio clínico?
- Participando en un estudio clínico, usted ayuda a la investigación médica. Así, usted contribuye al progreso de la medicina.
- Participando como paciente, usted disfruta de los tratamientos sanitarios más recientes y de un nuevo tratamiento potencial para su enfermedad, del que se investiga la eficacia y la seguridad.
- Los resultados de los estudios también pueden ayudar a futuros pacientes, mediante el desarrollo de un nuevo medicamento potencial, una nueva vacuna, una prueba diagnóstica mejor, etc.
- Es posible que el médico del estudio haga más pruebas de lo normal para evaluar el efecto del medicamento administrado de la mejor manera posible. No se cobrarán los gastos para estos tratamientos y/o pruebas adicionales que se realizan durante el estudio.
- Los estudios son estrictamente confidenciales y se garantiza su privacidad en todo momento.
Sin embargo, no se puede garantizar nunca que su participación le otorgue una ventaja personal. Usted también tiene que tomar en cuenta los esfuerzos adicionales y los posibles riesgos asociados con un estudio. Su médico del estudio siempre le explicará previamente y de forma detallada las posibles ventajas, desventajas y riesgos de un estudio específico.
¿Quién decide si usted puede participar en un estudio clínico?
El médico de la investigación decide si usted puede participar en un estudio clínico.
La participación en un estudio clínico es completamente voluntaria. Si usted decide no participar en el estudio, esta decisión no influirá, de ninguna manera, en su tratamiento o en la relación con su médico. Usted también puede decidir en cualquier momento si quiere dejar de participar en el estudio.
Encontrará más información sobre nuestro papel en nuestra política de referencia.
¿Qué pasa con sus datos durante un estudio clínico?
Si usted cumple los criterios de selección y decide participar en un estudio clínico, su participación, y todos sus datos personales y médicos, se tratarán de manera confidencial y de conformidad con las leyes locales. Durante un estudio clínico sus datos son recogidos y tratados de manera codificada. Estos datos son tratados y analizados, exclusivamente, en un contexto científico asociado con el estudio. En publicaciones científicas posteriores sobre los resultados del estudio, no se compartirá información de la cual se puede deducir la identidad de los participantes.
Su médico de investigación no compartirá, en ningún momento, con C-Lys los datos médicos que se obtengan durante un estudio clínico. En efecto, C-Lys solo interviene en la preselección de sujetos de ensayo candidatos.
Qué pasa si usted sufriera daños durante un estudio?
Cada estudio clínico se elabora cuidadosamente para minimizar los riesgos. Sin embargo, es imposible excluir problemas no previsibles. Por esta razón, la legislación establece que el promotor del estudio clínico tiene la obligación de contratar una póliza de seguro para la protección de los participantes. C-Lys solo interviene en la preselección de sujetos de ensayo candidatos y no es responsable por su participación en el estudio. Para más información sobre las pólizas de seguro, le remitimos al médico de investigación del estudio.
Si usted tiene interés en participar en un estudio clínico, un médico de investigación le puede explicar el objetivo del estudio y las posibles ventajas. Su médico también le señalará que estas ventajas no se pueden garantizar. Siempre pueden aparecer efectos secundarios debido a la fase de investigación del nuevo medicamento o tratamiento.
¿Usted recibe una compensación económica por su participación en un estudio clínico?
Generalmente, se prevé una compensación económica para voluntarios sanos que participan en un estudio de fase I. Ésta depende de la carga de las revisiones médicas (número de extracciones de sangre, pernoctaciones, etc.) que se realizan en el marco del estudio. Sin embargo, para la participación en estudios de fase II, III o IV la ley, normalmente, no permite dicha compensación. En algunos casos, usted tiene derecho a una cobertura de sus gastos de aparcamiento y desplazamiento. En ambos casos, el médico de investigación asociado con el estudio le podrá dar más información.
La compensación económica o una cobertura de gastos siempre proviene del promotor del estudio. Dado que C-Lys solo interviene en la preselección de sujetos de ensayo candidatos, C-Lys no es responsable, en ningún caso, del pago de una compensación a sujetos de ensayo.
¿Dónde debe comunicar los posibles efectos secundarios?
Cuando participa en un estudio clínico, resulta extremamente importante comunicar los posibles efectos no deseados o efectos secundarios a su médico. Así, ayudará a controlar la seguridad del medicamento o del tratamiento.
Este sitio web de C-Lys no es una plataforma para indicar los posibles efectos secundarios. Se aplica una regulación muy estricta para estudios clínicos con el fin de proteger a los participantes. Debido a esta situación jurídica única, los empleados de C-Lys no pueden responder a preguntas sobre productos médicos, tratamientos médicos, efectos secundarios o estudios clínicos específicos. Solo disponemos de la información que puede encontrar en este sitio web. La información sobre estudios específicos se ha elaborado consultando los investigadores y ha sido aprobada por una comisión ética autorizada.
Este sitio web de C-Lys tampoco es una plataforma para compartir sus experiencias con respecto a un estudio clínico concreto, por tentador que resulte compartir información con otras personas que participan en un determinado estudio clínico. Así, podría hablar sobre los posibles efectos secundarios o compartir sus experiencias, pero resulta muy importante que usted no lo haga. En efecto, puede influir en el resultado de la investigación, dado que existe la denominada fuerza de la sugestión: oír sobre un efecto secundario puede ser suficiente para que, efectivamente, ocurra.
Estudio clínico: de fase 1 hasta fase 4